发布时间:2022-11-29
11月29日,《Cell Discovery》期刊在线发表了题为“Loss of function of CMPK2 causes mitochondria deficiency and brain calcification”的研究论文,该研究由中国科学院脑科学与智能技术卓越创新中心熊志奇研究组与福建医科大学附属第一医院神经内科陈万金/王柠教授团队、山东大学齐鲁医院焉传祝/赵翠萍团队合作完成。该研究发现CMPK2基因双突变可导致线粒体功能障碍,并引起家族性脑钙化症(Familial Brain Calcification,FBC)。
脑钙化病是一种影响广泛的神经疾病,在老年人中发生率高达20%,由于其发病机制不清楚,临床上缺乏有效的治疗手段。FBC是一种以双侧基底节、小脑等脑区病理性钙化灶沉积为特征的神经遗传病,临床症状包括进行性运动、精神、认知障碍和头痛等。最常见的FBC包括原发性家族性脑钙化症(PFBC)、Aicardi-Goutières综合征(AGS),假性甲状旁腺功能减退症等。FBC的遗传基因鉴定对解析脑钙化发生的分子细胞机理非常重要。过去十年已经发现多个FBC特别是PFBC的关联基因,然而仍有40%以上的家族性患者的遗传因素尚未明确。
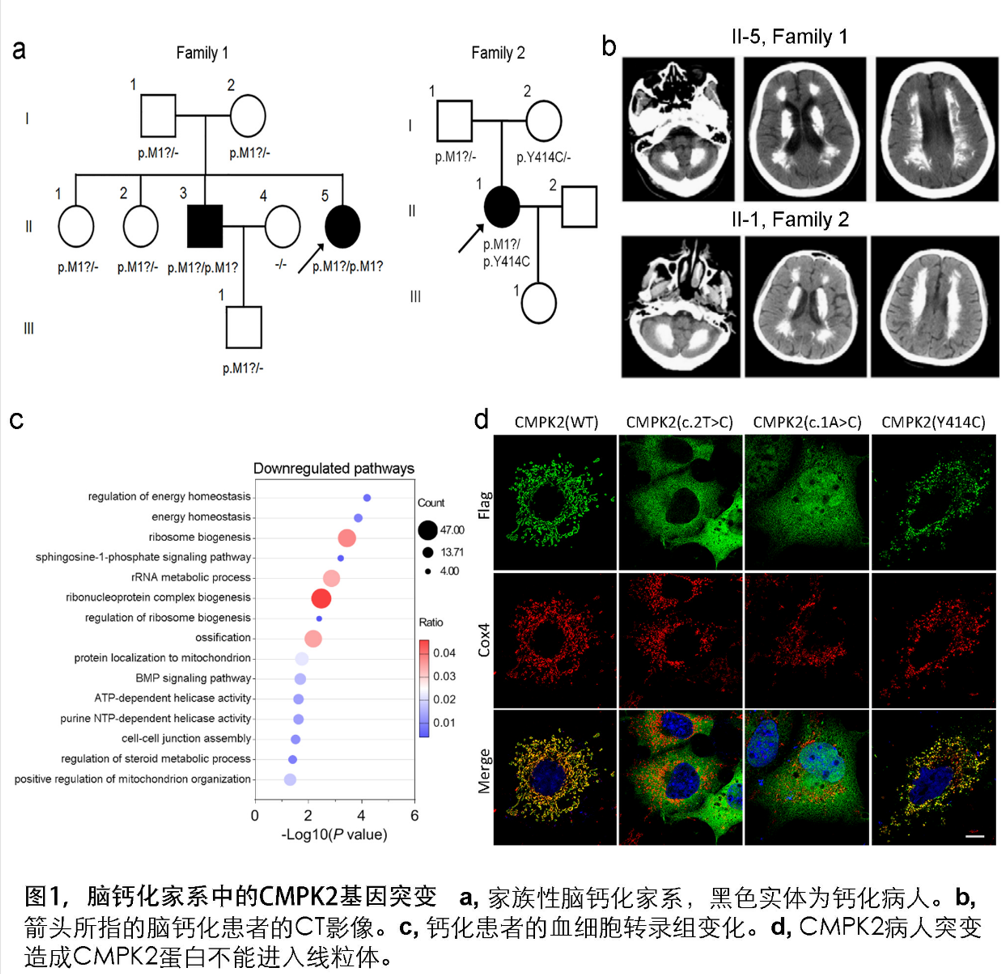
福建医科大学附属第一医院陈万金教授团队通过多年积累,建立了117个FBC家系和365个散发脑钙化患者的临床队列。运用全外显子测序技术,发现福建和山东的两个具有类似临床表征和脑钙化影像特征的家系患者均带有CMPK2基因的隐性突变或双杂合突变。进一步,采用Sanger测序验证了两个家系中的CMPK2基因突变与脑钙化符合共分离特征。CMPK2 基因编码一种定位于线粒体的UMP-CMP kinase 2 (CMPK2)蛋白质,是线粒体中负责将(d)CMP/UMP转化成(d)CDP/UDP关键酶。
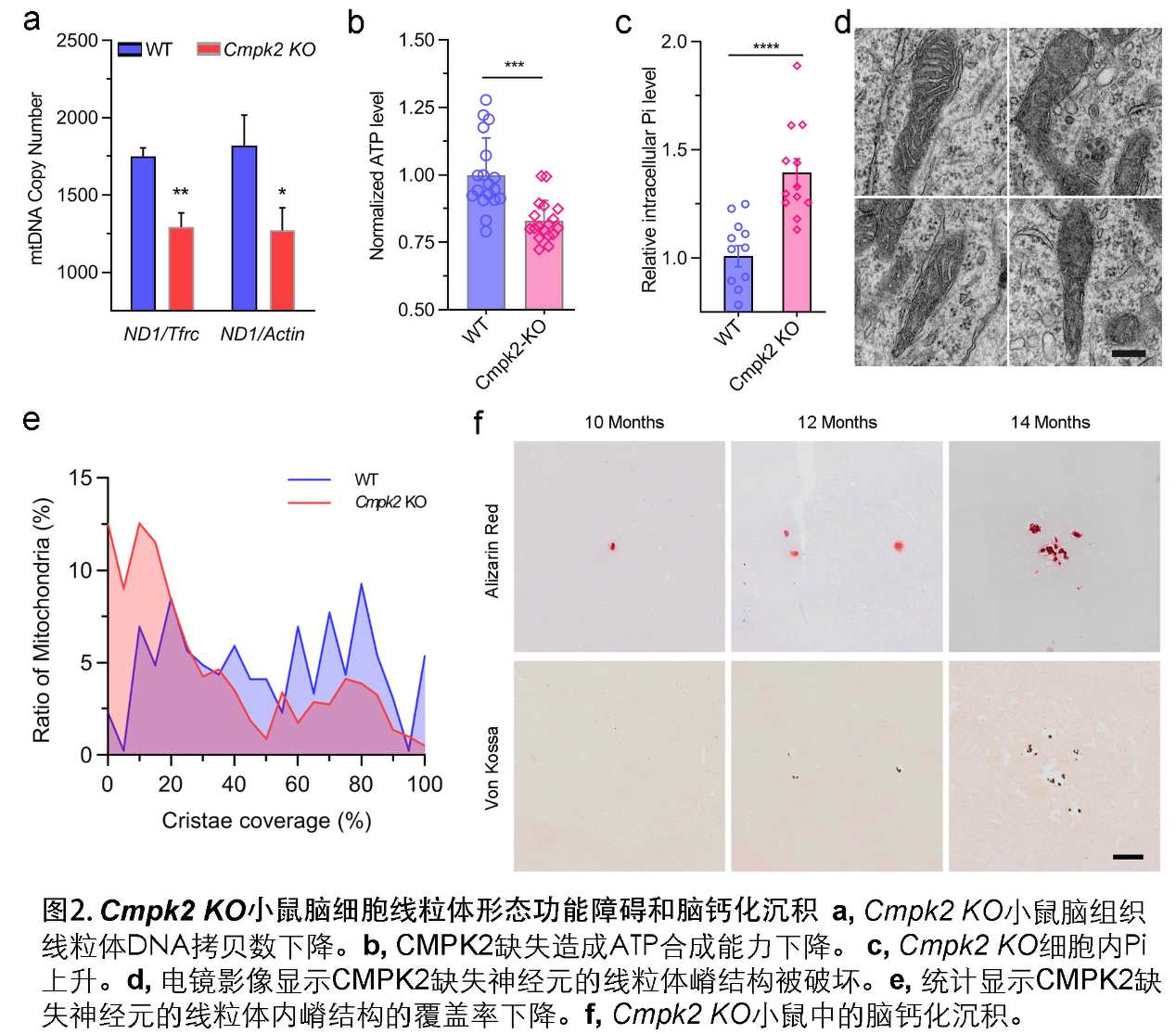
脑智卓越中心熊志奇研究组与陈万金团队合作,构建了突变蛋白质表达质粒,发现病人的突变造成CMPK2不能定位在线粒体。采集新鲜外周血提取血细胞RNA进行转录组测序,并采用基因功能注释的GO分析发现患者群体的差异性生物学通路出现多个线粒体和能量代谢相关通路的富集。利用原位杂交技术等表达分析技术发现,小鼠脑内的Cmpk2基因主要在神经元和血管内皮细胞呈现表达富集。Cmpk2敲除的小鼠脑组织线粒体DNA拷贝数减少。原代神经元培养分析显示,CMPK2缺失可造成线粒体关键蛋白如COX4, Cytochrome C的表达量降低,ATP合成减少以及细胞内无机磷酸根浓度增高。电镜观察显示,对线粒体功能至关重要的线粒体嵴结构在CMPK2缺失的细胞中被破坏。在钙化表型方面, Cmpk2基因敲除小鼠(Cmpk2-KO)以及带有患者突变的Cmpk2基因突变敲入(knockin, Cmpk2-KI)小鼠的丘脑区均出现年龄依赖的异常钙化沉积。
该研究证明了CMPK2基因突变是FBC的遗传致病因素之一,拓展了此前关于脑钙化发生的细胞学机制,即:CMPK2缺失引发线粒体核苷酸合成缺陷,并可能通过影响线粒体内的无机磷酸根稳态,导致脑钙化和多种临床症状的发生。
福建医科大学的陈万金、王柠和吴志英教授(现任浙江大学附属第二医院医学遗传科/罕见病诊治中心主任)与脑智卓越中心熊志奇研究组长期合作进行疾病基因的筛查与机理研究。在发作性运动诱发性运动障碍(PKD)领域,发现了首个致病基因PRRT2 (Nature Genetics, 2011)和第二个致病基因TMEM151A (Cell Discovery, 2021)。在家族性脑钙化病方向,发现了首个PFBC的隐性突变致病基因MYORG (Neuron, 2018),以及本次工作的CMPK2 (Cell Discovery, 2022)。2020年,双方团队在国际医学杂志BMJ发表分析文章,探讨单基因脑疾病的研究现状和未来展望(BMJ, 2020)。以新的致病基因为切入点,阐明发作性运动障碍和脑钙化等脑疾病发生的机理 (Cell Res, 2017; Cell Reports, 2021),有望为疾病的诊治带来突破性进展。
中科院脑智卓越中心光学成像平台、实验鼠房、电镜平台和非人灵长类研究平台,在疾病模型制作、饲养和成像方面提供了大力协助。该研究得到了国家自然科学基金委员会、中国科学院、科技部的资助。
 附件下载:
附件下载: 